

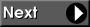
Obtain structure files for 1-methylcyclohexene, 3-methylcyclohexene, and methylenecyclohexane. You can easily sketch the chemical structures of the molecules using the online JME Editor and translate the resulting SMILES strings into 3D structures saved as MOL files. Each of the MOL files can be converted to PC GAMESS input with the program Open Babel: babel -i mol filename.mol -o inp filename.inp. Alternatively, you can use a molecular structure editor to build these molecules and save each geometry in a file.
Notice that building symmetric structures for cyclic systems such as methylenecyclohexane is not trivial and one often ends up with a molecule that is just slightly asymmetric. For example, the automatic conversion of JME editor output to PC GAMESS input gives a slightly asymmetric structure. Nearly symmetric molecular structures can be converted to perfectly symmetric molecular structures using a symmetrizer programs, such as SYMMOL by Tullio Pilati and Alessandra Forni. If you have compiled and installed SYMMOL, you can use the Python script mol_sym_gamess.py to convert the nearly symmetric structures in the MDL MOL format to the PC GAMESS input format. Running ./mol_sym_gamess filename.mol will produce files filename.sym and filename.uni containing coordinates for all and symmetry-unique atoms, respectively. If the symmetrization fails, the original coordinates in PC GAMESS input format are written to filename.geo.
If you want to continue with this tutorial without building the molecules, you may download the appropriate geometry specifications for 1-methylcyclohexene, 3-methylcyclohexene, and methylenecyclohexane. Notice that the latter file contains only the symmetry-unique atoms.
Next, the geometries of the three isomeric alkenes must be optimized. Because geometry optimization can be an expensive calculation, it is important to ask what kind of accuracy is required to answer the questions at hand. In many cases, one can optimize the geometry with a fairly modest basis set without losing much in accuracy. We will use the Hartree-Fock method with Pople's 6-31+G(d,p) basis set here due to speed considerations. Even though this approach suffers both from the lack of description of electron correlation and the limited size of the basis set, the errors from these two sources cancel to a good degree at this level of theory. When higher accuracy is needed, optimization at MP2/cc-pVTZ level could be used instead, but at much higher computational cost. In many systems, density functional theory also provides good geometries.
Add appropriate keywords to perform the HF/6-31+G(d,p) geometry optimization into each of the input files:
1-Methylcyclohexene and 3-methylcyclohexene are asymmetric molecules, thus the C1 label for the symmetry description. Notice that in the case of C1 symmetry, there is no blank line after the symmetry specification. Save these input files as CycHexe_1Met_HFO.inp, CycHexe_3Met_HFO.inp. Notice that we appended the HFO suffix to indicate that these are the Hartree-Fock optimization jobs. It is a good practice to use descriptive file labels because you are likely to generate several files for the same molecule during a real computational chemistry project. The input file for 1-methylcyclohexene should look like this:
$contrl scftyp=rhf runtyp=optimize nprint=-5 $end $system mwords=64 timlim=600 $end $basis gbasis=N31 ngauss=6 ndfunc=1 npfunc=1 diffsp=1 $end $statpt nstep=50 $end $data Tutorial: 1-methylcyclohexene HF/6-31+G(d,p) geometry optimization C1 C 6.0 2.48030 0.04020 -0.03980 C 6.0 0.97360 0.02580 -0.01680 C 6.0 0.35710 1.15990 0.07990 C 6.0 -1.13590 1.30420 0.12460 C 6.0 -1.80450 0.02270 -0.37780 C 6.0 -1.17320 -1.17110 0.34950 C 6.0 0.28110 -1.30200 -0.11340 H 1.0 2.83500 1.06830 0.03320 H 1.0 2.86210 -0.53650 0.80260 H 1.0 2.83370 -0.40070 -0.97190 H 1.0 0.95450 2.05810 0.13200 H 1.0 -1.44970 1.49720 1.15040 H 1.0 -1.43710 2.14030 -0.50660 H 1.0 -2.87280 0.05880 -0.16410 H 1.0 -1.64570 -0.07670 -1.45160 H 1.0 -1.20500 -1.00290 1.42600 H 1.0 -1.71890 -2.08150 0.10160 H 1.0 0.30160 -1.65240 -1.14540 H 1.0 0.79870 -2.02180 0.52070 $end
Methylenecyclohexane is a symmetric structure with one plane of symmetry (Cs). PC GAMESS allows several ways of defining symmetric structures. You can either specify positions of all the atoms and use coord=cart keyword in the $control group or specify only symmetry-unique atoms and use default (coord=unique) settings. If you were able to generate symmetric methylenecyclohexane structure, specify Cs for the symmetry label and include an empty line between the symmetry specification the molecular geometry specification. If you were not able to generate symmetric structure, you may optimize methylenecyclohexane in C1 symmetry the same way as other two alkenes. Save the file as CycHexa_Mete_HFO.inp. Your input with only symmetry-unique atoms should look like this:
$contrl scftyp=rhf runtyp=optimize nprint=-5 coord=unique $end $system mwords=64 timlim=600 $end $basis gbasis=N31 ngauss=6 ndfunc=1 npfunc=1 diffsp=1 $end $statpt nstep=50 $end $data Tutorial: methylenecyclohexene (Cs) HF/6-31+G(d,p) geometry optimization Cs C 6 0.330680 2.287200 0.000000 C 6 -0.119710 1.057100 0.000000 C 6 -0.383460 0.327600 1.298650 C 6 0.325890 -1.027350 1.251550 C 6 -0.113600 -1.789000 0.000000 H 1 0.518480 2.793510 -0.935300 H 1 -1.456110 0.175300 1.418550 H 1 0.001440 0.913550 2.133250 H 1 0.066500 -1.605200 2.138600 H 1 1.404190 -0.870790 1.221900 H 1 -1.198900 -1.890200 0.000000 H 1 0.341600 -2.779300 0.000000 $end
Submit your calculations. Under Linux, specify the path to PC GAMESS libraries with the -ex flag. For example, to start the optimization of 1-methylcyclohexene, navigate into a directory with sufficient free disk space and enter pcgamess -i CycHexene_1Met_HFO.inp -o CycHexene_1Met_HFO.out -ex /usr/local/pcgamess & at the Unix shell. The calculation will take some time and unless you are working on a multiprocessor system, it would be better if you wait until the first calculation finishes before the next one is started. You can monitor the energy convergence during the optimization using the Unix command grep FINAL CycHexene_1Met_HFO.out. When the calculation is finished, you can delete any temporary files such as DICTNRY, AOINTS. Rename the file PUNCH into something more descriptive, such as CycHexe_1Met_HFO.pun.
Examine the output text files CycHexene_1Met_HFO.out, CycHexene_3Met_HFO.out and CycHexane_Mete_HFO.out to verify that the equilibrium geometry was successfully located. Notice that each optimization required a large number (nearly 40) of steps before the equilibrium geometry was located. Luckily the calculation of each step was fast because of the small number of the basis functions in this system. Notice that the time required for the optimization of each of the asymmetric alkenes (ca. 1 hour on a modern 2 GHz processor) is nearly twice as long as the time required to optimize the symmetric alkene. The difference arises mainly from the fact that the SCF energy and derivatives are nearly twice as fast in Cs symmetry than in C1 symmetry for the same number of atoms and basis functions. Thus, for the best performance, you should take advantage of the symmetry whenever possible.
Quantum chemical calculations of molecules are necessarily approximate and thus inaccurate. In general, more accurate methods are more time-consuming. This makes highly accurate methods ill-suited for geometry optimizations. Furthermore, efficient geometry optimization requires analytical energy gradients, which may not be possible with more accurate methods. For example, PC GAMESS only supports analytical energy gradients at the Hartree-Fock level. Thus, geometry optimizations are often performed with modest basis sets ignoring dynamic electron correlation.
It is common to obtain a better estimate for the energy of the optimized structure by performing a single point calculation with a more accurate method using the structure optimized at the lower level. The single point calculation typically will include some treatment of electron correlation and may employ a larger basis. We will next perform MP4(SDQ)/6-31+G(d,p) calculation on the HF/6-31+G(d,p) optimized geometry to get a more accurate energy value for this structure.
The PC GAMESS output file provides the coordinates of the optimized geometry just below the statement EQUILIBRIUM GEOMETRY LOCATED. You can copy and paste these coordinates into a new input file and use the keywords runtyp=energy mplevl=4 to enable MP4(SDQ) single point calculation. For symmetric molecules, use coordinates of symmetry unique atoms. The input for 1-methylcyclohexene should look like this:
$contrl scftyp=rhf runtyp=energy mplevl=4 nprint=-5 $end $system mwords=64 timlim=600 $end $basis gbasis=N31 ngauss=6 ndfunc=1 npfunc=1 diffsp=1 $end $data Tutorial: 1-methylcyclohexene MP4/6-31+G(d,p)//HF/6-31+G(d,p) energy C1 C 6.0 2.5156121796 0.0097992136 -0.0478553419 C 6.0 1.0101937015 0.0643618931 -0.0030230606 C 6.0 0.3428259858 1.2065331526 0.0925053956 C 6.0 -1.1624809168 1.3187640500 0.1122780494 C 6.0 -1.8414757614 0.0266139732 -0.3446461922 C 6.0 -1.1766316408 -1.1852467814 0.3049728520 C 6.0 0.2997956685 -1.2705116813 -0.0856737553 H 1.0 2.9537511219 1.0010586319 -0.0192883313 H 1.0 2.9075522747 -0.5603693151 0.7914705619 H 1.0 2.8563192951 -0.4853305039 -0.9549260789 H 1.0 0.8954064452 2.1309908013 0.1619219600 H 1.0 -1.4918494552 1.5754075352 1.1194481183 H 1.0 -1.4704399743 2.1468473381 -0.5220392455 H 1.0 -2.9013754299 0.0557009714 -0.1059114844 H 1.0 -1.7638104002 -0.0590633698 -1.4271628001 H 1.0 -1.2586119892 -1.0997419156 1.3871619564 H 1.0 -1.6881584707 -2.1014134583 0.0220760686 H 1.0 0.3962737937 -1.6566989292 -1.1011620809 H 1.0 0.8120035726 -1.9878016059 0.5529534089 $end
Save the input files as CycHexe_1Met_MP4.inp, CycHexe_3Met_MP4.inp, and CycHexa_Mete_MP4.inp. Notice that we appended the MP4 suffix to indicate that this is the fourth-order Moller-Plesset job. Submit your calculations and examine the output files. The calculation will be faster than the optimization job. However, because of the advanced treatment of electron correlation, an MP4(SDQ) step takes about six times longer than the Hartree-Fock step with this basis. Again, the calculation on the symmetric structure is significantly faster. If you wish to continue without actually performing the calculations, you may download the output files for 1-methylcyclohexene, 3-methylcyclohexene, and methylenecyclohexane MP4 calculations.
Locate the section starting with RESULTS OF MOLLER-PLESSET 4TH ORDER CORRECTION ARE in the MP4 output files. Record the RHF, MP2, MP3, and MP4 absolute energies in a table. Your table of results should look something like this:
| MOLECULE | ABSOLUTE ENERGIES, IN HARTREE | ||||
|---|---|---|---|---|---|
| HF | MP2 | MP3 | MP4 | ||
| 1-methylcyclohexene |
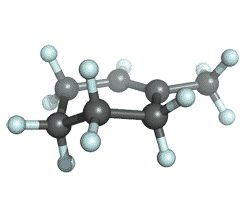 |
-272.080994 | -273.071301 | -273.135338 | -273.144439 |
| 3-methylcyclohexene |
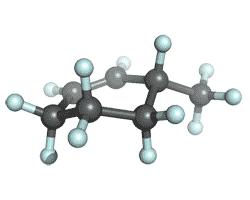 |
-272.077965 | -273.068416 | -273.132495 | -273.141561 |
| methylenecyclohexane |
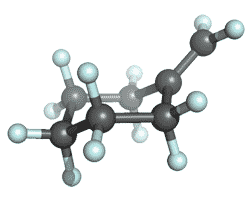 |
-272.078164 | -273.068528 | -273.132027 | -273.141618 |
A glance at the table indicates that 1-methylcyclohexene is predicted to be the most stable alkene. Typically, one is interested in relative energies of the structures with respect to the lowest energy structure. The relative energies are typically expressed in kJ/mol, kcal/mol, eV, or cm-1 units. For example, the relative energy of 3-methylcyclohexene at the Hartree-Fock level in kcal/mol units can be evaluated as 627.51*(272.080994-272.077965) = 1.90 kcal/mol. Prepare a table showing the relative energies of 3-methylcyclohexene and methylenecyclohexane. Your table (in kcal/mol units) should look something like this:
| RELATIVE ENERGIES, IN KCAL/MOL | ||||
|---|---|---|---|---|
| HF | MP2 | MP3 | MP4 | |
| 1-methylcyclohexene | 0.00 | 0.00 | 0.00 | 0.00 |
| 3-methylcyclohexene | 1.90 | 1.81 | 1.78 | 1.81 |
| methylenecyclohexane | 1.78 | 1.74 | 2.07 | 1.77 |
Notice that all the methods predict that 3-methylcyclohexene and methylenecyclohexane are about 2 kcal/mol less stable than 1-methylcyclohexene. This is consistent with the thesis that alkenes with most substituents at the sp2 carbon are thermodynamically the most stable.