

Prepare the structures of the two carbocation intermediates using a full-fledged molecular structure editor, such as MOLDEN. This time the JME Editor is not as helpful because of the difficulty of specifying the cationic carbon. You can build the carbocations using the optimized structures of alkenes as starting points. For example, the structure of the secondary carbocation can be readily obtained by adding a hydrogen atom to the optimized 3-methylcyclohexene. The structure of the tertiary carbocation can be readily obtained by adding a hydrogen atom to the optimized methylenecyclohexane because these two molecules have a similar symmetry. Make sure to specify a tetrahedral geometry that does not break the CS symmetry when converting the terminal methylene to a methyl group. Write the structures out as MOL files files and use mol_sym_gamess script to convert these into the PC GAMESS input format.
If you want to continue with this tutorial without building the molecules, you may download the appropriate geometry specifications for secondary carbocation, and tertiary carbocation. Notice that the latter file contains only the symmetry-unique atoms.
Add appropriate keywords to perform HF/6-31+G(d,p) geometry optimization. The basic input is the same as for alkene optimization with a few differences. First, a charge of +1 should be specified for carbocations. Second, we request that the second derivative matrix (hessian) is evaluated at the end of the calculation to ensure that the structure is indeed the energy minimum. Such validation is a good practice when performing quantum chemical optimizations but is especially important when dealing with unstable species such as carbocation intermediates. The hessian calculation can distinguish a true minimum (e.g. the carbocation) from other stationary points (e.g. transition state) that an optimizer might have found. A true minimum will have all hessian eigenvalues positive. Edit the input files to add keywords for the optimization job and add the following:
If you were able to symmetrize the tertiary carbocation, your input file for this molecule in Cs symmetry should look like shown below. If you were unable to use a symmetrizer program, you can learn here how to manually symmetrize nearly-symmetric structures in XYZ coordinates.
$contrl scftyp=rhf runtyp=optimize coord=unique icharg=+1 nprint=-5 $end $system mwords=64 timlim=600 $end $basis gbasis=N31 ngauss=6 ndfunc=1 npfunc=1 diffsp=1 $end $statpt nstep=70 hssend=.t. $end $data Tutorial: tertiary carbocation HF/6-31+G(d,p) geometry optimization Cs C 6 0.337000 -0.401000 1.266000 C 6 1.102716 -0.102376 0.000000 C 6 -1.827630 -0.048194 0.000000 C 6 -1.034000 0.289000 1.265000 C 6 2.327449 0.402001 0.000000 H 1 3.198406 -0.231123 0.000000 H 1 2.675000 1.040000 0.800000 H 1 -2.071308 -1.110447 0.000000 H 1 -2.773567 0.487422 0.000000 H 1 -1.594000 0.003000 2.150000 H 1 -0.890000 1.366000 1.324000 H 1 0.913000 -0.101000 2.138000 H 1 0.186000 -1.480000 1.340000 $end
Save the input files as CycHex_SecCat_HFO.inp and CycHex_SecCat_Ter_HFO.inp. Submit the first calculation; the optimization will take some time. You can monitor the progress of optimization visually using MOLDEN. However, PC GAMESS, when running under Linux, produces DOS-type text files that have different end-of-line characters than Unix text files. Thus, when examining PC GAMESS output from a running job, you should convert the output into the Unix format using the Unix command dos2unix -n CycHex_SecCat_HFO.out CycHex_SecCat_HFO.unx and then examine the Unix file with MOLDEN. Clicking on the Geom. Conv. button in MOLDEN allows to see the convergence of energy and change in forces. When the calculation is finished, you can delete any temporary files such as DICTNRY, AOINTS as well as the converted output file CycHex_SecCat_HFO.unx. Rename the file PUNCH into something more descriptive, such as CycHex_SecCat_HFO.pun. Submit the second calculation.
Examine the output text files CycHex_SecCat_HFO.out and CycHex_TerCat_HFO.out to verify that the equilibrium geometry was successfully located. Also, verify that all vibrational frequencies are positive; ignore the first six frequencies which correspond to translations and rotations. These six frequencies should be nearly zero for well-converged geometry. Notice that each optimization again required a large number (nearly 50) of steps before the equilibrium geometry was located. Notice that the number of optimization steps was roughly the same for symmetric and asymmetric structures but the tertiary carbocation calculation finished faster because each step took less time.
The vibrational analysis in the PC GAMESS output files can be visualized with the programs MOLDEN and MOLEKEL. You can safely convert the output file of a finished job into the Unix format by running dos2unix CycHex_SecCat_HFO.out; this will simply create file CycHex_SecCat_HFO.out in Unix format. To visualize molecular vibrations from the file that contains both optimization and hessian calculations, use -H flag when starting molden: molden -H CycHex_SecCat_HFO.out, then click on Freq button to bring up a window that allows visualization of individual normal modes.
PC GAMESS provides some facilities to speed up single-point energy calculations calculations after geometry optimization. Each PC GAMESS calculation will create a file named PUNCH which will include information about the final optimized geometry, molecular orbitals, and if the hession calculation was requested, the hessian and vibrational modes. A single point calculation can be started using the final geometry and molecular orbital coefficient from this file. It is easiest to reuse the molecular orbital coefficients if the basis set of the optimization job and single point energy are the same. Similarly, a new higher-level optimization can be started from the final geometry and the saved hessian matrix.
A new input file can be generated by copying the converged geometry, molecular orbitals, and hessian from the existing PUNCH file. This process can be automated, for example a small Python script parse_punch.py will create relevant parts of the new input file based on the existing PUNCH file. The beginning of an input file that requests single point MP4/6-31+G(d,p) calculation based on stored MO's is shown below. Notice how the guess group keywords now specify that existing 198 molecular orbitals must be used to form the initial guess.
$contrl scftyp=rhf runtyp=energy mplevl=4 coord=unique icharg=1 nprint=-5 $end $system mwords=128 timlim=600 $end $basis gbasis=N31 ngauss=6 ndfunc=1 npfunc=1 diffsp=1 $end $guess guess=moread norb=198 $end $data Tutorial: tertiary carbocation HF/6-31+G(d,p) geometry optimization CS 0 C 6.0 0.3725395583 -0.5642825158 1.2532286922 C 6.0 1.0429018926 -0.1912292973 0.0000000000 C 6.0 -1.7719493228 0.0396900864 0.0000000000 C 6.0 -0.9487585629 0.2924240164 1.2615058297 C 6.0 2.2547123239 0.6407512817 0.0000000000 H 1.0 3.0717463311 -0.0904965997 0.0000000000 H 1.0 2.3578638884 1.2356964754 0.8974985249 H 1.0 -2.1447789379 -0.9807557922 0.0000000000 H 1.0 -2.6386092415 0.6915828898 0.0000000000 H 1.0 -1.4900382873 0.0170335957 2.1575187089 H 1.0 -0.6977004600 1.3450003452 1.3530934343 H 1.0 0.9761248556 -0.3538222604 2.1250949174 H 1.0 0.0939904851 -1.6121794408 1.2357910143 $end $VEC 1 1 3.22740869E-03 2.78099393E-04 6.81259795E-05 1.39065891E-05 5.71032683E-05 1 2 3.41605856E-03 7.23503846E-04-8.44041476E-05 1.01930302E-03 1.27319098E-03
Submit your calculations and examine the output files. If you wish to continue without actually performing the calculations, you may download the output files for the secondary and tertiary carbocations. First, notice that it took only a few seconds to calculate the Hartree-Fock energy when the initial guess orbitals were given. Second, notice that the time needed for the MP4 calculation of the symmetric carbocation is about half of the time needed for asymmetric carbocation. For example, running the tertiary carbocation calculation with C1 symmetry without reading in the optimized orbitals as initial guess required 6.7 minutes versus 2.9 minutes for symmetric structure with initial guess. Reading in the optimized orbitals allows for a significant saving in computational time when a large number of basis functions are used.
Locate the section starting with RESULTS OF MOLLER-PLESSET 4TH ORDER CORRECTION ARE in the MP4 output files. Record the RHF, MP2, MP3, and MP4 absolute energies in a table. Your table of results should look something like this:
| MOLECULE | ABSOLUTE ENERGIES, IN HARTREE | ||||
|---|---|---|---|---|---|
| HF | MP2 | MP3 | MP4 | ||
| Tertiary Carbocation |
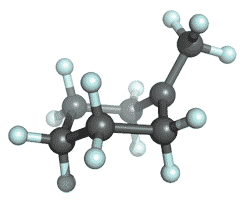 |
-272.4175234 | -273.3915509 | -273.4602420 | -273.4701929 |
| Secondary Carbocation |
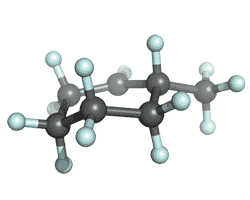 |
-272.3960185 | -273.3664015 | -273.4368879 | -273.4466425 |
A glance at the table indicates that tertiary carbocation is predicted to be more stable than the secondary carbocation. The relative energy of the secondary carbocation can be obtained as the energy difference, multiplied with a conversion factor to get commonly used units such as kcal/mol. For example, the relative energy of the secondary carbocation at the Hartree-Fock level in kcal/mol units can be evaluated as 627.51*(272.4175234-272.3960185) = 13.49 kcal/mol. Prepare a table showing the relative energies of secondary carbocation with different methods. Your table (in kcal/mol units) should look something like this:
| RELATIVE ENERGY, IN KCAL/MOL | ||||
|---|---|---|---|---|
| HF | MP2 | MP3 | MP4 | |
| secondary carbocation | 13.49 | 15.78 | 14.65 | 14.78 |
Notice that all the methods predict that the tertiary carbocation is more stable by about 13-16 kcal/mol. This kind of spread of values with different methods is not uncommon; typically the highest-level (in this case MP4) energy is considered the most accurate. The higher thermodynamic stability of tertiary carbocation is consistent with the thesis that the primary carbocation can rearrange into tertiary carbocation, which will then yield 1-methylcyclohexene and methylenecyclohexane.