

Semiempirical calculations allow to the study large molecular systems and thus are of considerable interest to practicing organic chemists who are concerned about the energetics of reactions with larger molecules. Semiempirical approaches can also be used to model molecular complexes such as between a reactant and catalysts and thus may shed light into designing optimal catalysts for a given reaction. Semiempirical methods have been also employed in the studies of enzyme-catalyzed reactions via QM/MM approach in which the substrate and the active site residues are modeled via semiempirical methods while the surrounding protein and solvent water are treated by a molecular mechanics force field.
Setting up a semiempirical calculation is fairly straightforward. Most commercial programs offer a graphical user interface for launching the calculation. You build the molecule, specify its charge and spin state, choose a semiempirical method (MNDO, AM1, PM3 ...) and launch the calculation. Such programs do a good amount of deciding for you; for example they choose internal versus a Cartesian coordinate system for the optimization. In some cases you can specify the optimization algorithm, e.g. full Newton-Rhapson, or BFGS.
Quantum chemical calculations generally require the following elements in the input:
Further optional directives are used to control various aspects of calculation
In this course we will learn to use a freely available program PC GAMESS / Firefly. This program is harder to use, but offers excellent speed, and you can run it at your home under the Windows operating system. One of the difficulties in using academically oriented programs such as PC GAMESS / Firefly stems from the academic freedom they provide: the user has a great control over every aspect of calculation. Exercising this control, however, requires a knowledge of many kewords that are specific to this program. You will learn a small subset of these keywords in this course. The PC GAMESS / Firefly website offers some information on how to run this program; a full manual for a related program GAMESS is available here
PC GAMESS also requires that the user defines molecular symmetry. The concept of molecular symmetry is intuitively familiar to chemists and helps significantly to speed up calculations. However, the creation of input that incorporates molecular symmetry requires practice and we will consider all molecules as lacking symmetry in this introductory tutorial. Molecules that lack symmetry belong to the C1 symmetry group.
PC GAMESS input consists of a header part that defines the keywords and options that control the calculation. The keywords are organized into logical groups such as $control to control the overall flow of the program, or $statpt to control how the geometry optimization is carried out. After the header comes a $data group that starts with a title line, followed by one or two lines for definition of molecular symmetry, followed by the specification of molecular geometry. Each group starts with the group name and ends with the $end statement. For example, the data group should have $data on a line before the molecular data begins and $end at the end. It is a good style to start group definitions at position 2. Below is an example layout of the PC GAMESS (Firefly) input file:
$contrl runtyp=optimize nprint=-5 coord=zmt $end $basis gbasis=pm3 $end $statpt method=nr hess=calc ihrep=0 $end $data TITLE GOES HERE C1 Z-MATRIX GOES HERE $end
The performance of various computational methods is often tested by calculating enthalpies of isomerization reactions. Such enthalpies are readily available experimentally for the gas phase isomerization reactions because they can be calculated from standard enthalpies of formation of two molecules. The paper Performance of B3LYP Density Functional Methods for a Large Set of Organic Molecule presents one such analysis. Consider the reaction 8 in Table 3 and then build structures of cyclopentene and vinylcyclopropane with MOLDEN. While in the Z-matrix editor, save each structure out in GAMESS-US Z-matrix format. Edit the input file by adding the appropriate control structures to carry out the Newton-Rhapson geometry optimization with PM3 method. Delete the zmat angstroms line near the beginning, and constants and end statements from the z-matrix. Also, erase the text variables but leave an empty line in its place. (i.e. there should be an empty line after z-matrix setup and before the variable value assignments). The result should look like this. Save your file as filename.inp.
Run the calculation by typing firefly -i filename.inp -o filename.out. When the calculation has finished type dos2unix filename.out. Then examine the result with MOLDEN by issuing molden filename.out
The dehydration of substituted alcohols produces a mixture of isomeric alkenes. The product ratios in such an elimination reaction are determined largely by the thermodynamic stability (heat of formation) of the carbocation intermediates and the final alkenes. The kinetic barriers (transition states) play a secondary role in determining the observed product ratios. The following tutorial illustrates how semiempirical methods can be used to predict or to rationalize the outcomes of chemical reactions when the product ratios are determined by their thermodynamic stability or by the stability.
Draw a reaction mechanism that would rationalize the formation of 1-methylcyclohexene, 3-methylcyclohexene, and methylenecyclohexane when 2-methylcyclohexanol is dehydrated in the presence of a strong acid.
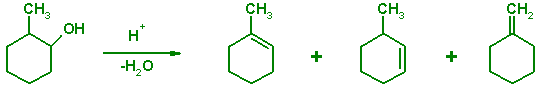
Prepare the structures of the two carbocation intermediates using the Z-Matrix Editor in the program MOLDEN. An easy way to build the required structures is to build the hydrogen molecule (Add Line) first, then replace one of the hydrogens with the cyclohexyl substituent to give cyclohexane. The structure of a carbocation can be obtained by substituting one of the hydrogens with the methyl group and removing (Delete Line) a hydrogen atom at the cationic center. Because you will be performing quantum chemical calculations, you do not need to worry about assigning partial charges to the atoms in the carbocation. However, your optimization will finish faster if you alter the bond distances and angles in the Z-matrix so that they are close to the actual structure of the carbocation (e.g. planar trigonal geometry at the cationic center). Save the structures as GAMESS-US Z-matrices naming them sec_carbo.inp and ter_carbo.inp, for example. Notice that the molecule names are arbitrary; the extension inp is commonly used for GAMESS input files. Optimize the structures with PC GAMESS / Firefly as done previously and recond the final energies. Are the results consistent with your chemical intuition about stabilities of carbocations and substituted alkenes?