
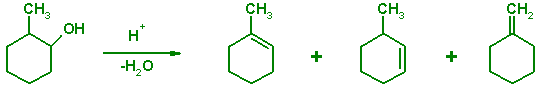
Dehydration of substituted alcohols produces a mixture of isomeric alkenes. The product ratios in such elimination reaction are determined largely by the thermodynamic stability (heat of formation) of the carbocation intermediates and the final alkenes. The kinetic barriers (transition states) play a secondary role in determining the observed product ratios. The following tutorial illustrates how semiempirical methods can be used to predict or rationalize outcomes of chemical reactions when the product ratios are determined by their thermodynamic stability or by the stability.
Draw a reaction mechanism that would rationalize the formation of 1-methylcyclohexene, 3-methylcyclohexene, and methylenecyclohexane when 2-methylcyclohexanol is dehydrated in the presence of a strong acid.
Prepare structures of the two carbocation intermediates using the Z-Matrix Editor in the program MOLDEN. An easy way to build the required structures is to build the hydrogen molecule (Add Line) first, then replace one of the hydrogens with the cyclohexyl substituent to give cyclohexane. The structure of a carbocation can be obtained by substituting one of the hydrogens with the methyl group and removing (Delete Line) an hydrogen atom at the cationic center. Because you will be performing quantum chemical calculations, you do not need to worry about assigning partial charges to atoms in the carbocation. However, your optimization will finish faster if you alter the bond distances and angles in the Z-matrix such that they are close to the actual structure of the carbocation (e.g. planar trigonal geometry at the cationic center). Save the structures as Gaussian Z-matrices naming them sec_carbo.dat and ter_carbo.dat, for example. Notice that the molecule names are arbitrary but extensions such as inp, dat, com are commonly used for Gaussian input files.
You will be using a computational chemistry program Gaussian03 to perform the calculations on carbocations. Gaussian is one of the most commonly used computational chemistry programs and it is important that you learn well how to use it. One valuable resource is their on-line technical documentation. Edit your sec_carbo.dat file with a text editor to append the following at the beginning of the file:
%Mem=16MW # PM3 Opt=CalcAll Secondary carbocation: PM3 Newton-Rhapson Minimization 1 1
Inspect your file. A typical Gaussian input file will have a section that allocates computational resources (Link 0 commands that start with %) followed by the route section (lines that start with #), the title section (free format text), and molecule specification. There are three blank lines: one separating the route section from the title section, the other separating the title section from the molecule specifications, and the last one at the end of the molecule specification. The section that you added tells the Gaussian program to allocate 16 megawords (128 MB) of memory and perform optimization while evaluating the Hessian at each point. It also specifies the charge (1) and spin multiplicity (1) for the molecule. Both the charge and the spin multiplicity are integers. Charge is the total charge of the system (e.g. zero for a zwitterionic amino acid) and the multiplicity is 1 for closed-shell molecules in the ground electronic state. Multiplicity is 2 for single radicals and 3 for diradicals, such as molecular oxygen.
Submit your calculation to the queue using the subg03 command.
Edit the second file and change the comment line to read "Tertiary carbocation" this time and submit this job as well. Inspect the text of the output. This file can be rather long because of many minimization steps. You are interested in energy (not the free energy) at the minimum, which would be the last step. Gaussian reports energy values (Energy= ) in the Hartree units (1 Hartree = 627.51 kcal/mol). You can use the UNIX command grep to display relevant lines: grep 'Energy= ' filename. Alternatively, you can use MOLDEN to visualize the structure and the course of optimization; holding your mouse over the point will display the Hartree-Fock SCF energy. Convert the heats of formation of minimized structures from hartrees to kcal/mol and report these heats of formation.
Prepare structures of the three stable products using MOLDEN and perform an PM3 optimization on each structure using Gaussian 03. Record the heats of formation for optimized structures.
Consider the dehydration of cis-2-methylcyclohexanol. Compare the stability of the three alkene products products with the AM1, PM3 and MNDO models. Compare the results with experimentally available heat of isomerization between 1-methylcyclohexene and 3-methylhexene. The gas phase thermochemistry data for many molecules is available from the NIST Chemistry Webbook. Calculate the heat of formation of the starting compound and evaluate heats of reactions leading to the three alkenes. Compare the calculated heats of formations and heats of reactions with experimental thermochemical data, when available. Which semiempirical method appears most reliable?
Repeat the calculations with AM1, PM3, and MNDO model for the two carbocations. Find from the literature the observed ratios of three alkene products and discuss if the semiempirical calculations allow prediction of reactivity in this system. Which empirical organic chemical rule can be rationalized based on these calculations?
The following assignment is based on a real-life application of semiempirical methods and was contributed by Dr. Bowlus who studied the problem in 1986 while working with SDS Biotech Corporation. The synthesis of a insecticide 3 was hampered because the oximation of the geminal dichloride 1 with hydroxylamine into a desired synthetic intermediate 2 produced side-products that were difficult to characterize experimentally. A working hypothesis was that a stereoisomer arising from E/Z isomerization of the oxime moiety formed. The E-stereoisomer could have subsequently reacted with the nearby nitrile group to give a five-membered ring. A counter argument for this hypothesis was based on the expected high steric cost of formation of a five-membered ring in this already strained system.
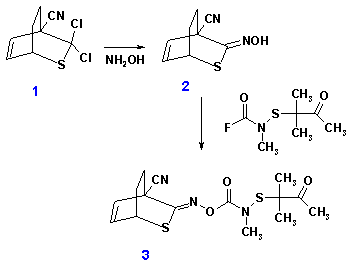
Perform conformational analysis of compound 2 with the MNDO model to see if the stereoisomer that could undergo the ring closure is stable enough to form. You need to build a fairly good starting structure and may need to use Newton-Rhapson minimization in order to converge to a minimum; the minimization may take a large number of steps if the starting structure is not very good. What is the reaction enthalpy for the ring closure reaction? Is the counter-argument about the excessive strain (say more than 5 kcal/mol) valid? You can use a program of your choice to perform these calculations.
Solve the Level 2 problem also using the PGGD/PM3 or PGGD/MNDO model as implemented in the program BOSS. Perform additional semi-empirical calculations to identify another possible side-product that could form when excess hydroxylamine reacts with the geminal dichloride 1. You can use the program MOLDEN to read in optimized MNDO structures and write out files that can be converted to BOSS Z-matrix input using the program autozmat. The PGGD/PM3 optimization and energy evaluation of neutral molecules can be performed with the xPDGOPT script. The command xPDGOPT input will perform PDDG/PM3 optimization given the file input.z.