Chemical reactions occur by the rearrangement of nuclear configurations from the reactant state to the product state. For polyatomic molecules, there is an enormously large number of possible rearrangement paths that take reactants to products. Reactant molecules that have lots of energy could follow a path that involves high energy configurations, reactants with less energy will follow a path that involves configurations with lower energy. A complete description of a chemical reaction dynamics would include all these paths. However, such a complete description is challenging because of the need to map out a multidimensional potential energy surface. Instead, a simplified approach, termed the transition state theory, is commonly employed.
The simplest versions of the transition state theory assume that reactants behave like very tired mountain climbers who are trying to get from one valley to another and have to cross a mountain range. Such mountain climbers will seek out the easiest path, one that avoids steep climbs and raises minimally in altitude. They will seek out a gorge that takes them over the ridge. Like a group of tired mountain climbers, the reactant molecules in the transition state theory will follow a unique path that connects the reactant basin and the product basin. The highest point on this path is the col, or saddle point that separates the reactant basin from the product basin. The saddle point is the point of highest energy along the reaction path and is also the point of lowest energy in the direction perpendicular to the reaction path (lowest point of the ridge that separates reactants and products.
Transition states correspond to saddle points on the potential energy surface. Like minima, the saddle points are stationary points with all forces zero. Unlike minima, one of the second derivatives in the first order saddle is negative. The eigenvector with the negative eigenvalue corresponds to the reaction coordinate. Transition state search thus attempts to locate stationary points with one negative second derivative. The basic recipe is: identify the reaction mode and maximize energy along this mode while minimizing energy in all other directions. One reason why transition state optimization is more difficult than the search for a minimum is that a successful search should start off in a region where the reaction coordinate already has a negative curvature. In other words, search for a transition state should start near the transition state.
Here are some approaches to locate transition state structures for chemical reactions:
You can use MOLDEN to build the reactant, product, and the transition state structures and wrote these out as text files. You can use bluefish to add the headers in order to run MP2/aug-cc-pVTZ optimization and transition state search calculations. You can follow the first approach (optimization based on second derivatives) to locate the transition state. Below are the input and output files so you can check your works:
The enery of the optimized molecule is saved in the output file. In Linux system, one gould use the grep command to search for lines that contain the desired values. For example, MP2 energy from Gaussian calculation of HCN can be found by issuing grep EUPM2 HCN_aTZ_MP2_Opt.out command. Here is the summary of energies:
The energy difference between the product (hydrogen isocyanide) and reactant (hydrogen cyanide) gives the reaction energy ΔE. The energy difference between the transition state and the reactant gives the activation energy Eact. Quantum mechanical programs usually give energy values are in atomic units. Conversion from atomic units (Hartree) to SI units (kJ/mol) involves multiplication with a factor 2625.50.
One of the first things to do after a successful geometry optimization is to calculate vibrational frequencies. The vibrational frequencies are related to second derivatives; a minimum will have only positive frequencis while transition state should have one negative frequency. The vibrational analysis must be performed at the the optimized geometry. Gaussian saves the optimized geometry into a checkpoint file, and the geometry can be recalled from this file before performing the vibrational analysis. The input and output files for vibrational analysis for HCN isomerization are shown below:
We used MOLDEN to briefly look at the vibrations in the transition state and saw that the negative frequency corresponded to the motion of hydrogen between carbon and nitrogen. Vibrations of the reactant (HCN) and product (HNC) can be also visualized; these vibrations correspond to spectras lines in the infrared spectra of molecules. If your browser is modern enough to support HTML5 video, you can play back the animations of molecular vibrations of the hydrogen atom in the reactant, product, and the transition state.
As part of the vibrational analysis, the enthalpies and Gibbs free energies and are evaluated based on the molecular mass, molecular geometry and vibrational frequencies. Also, breakdown of thermal energy and entropy among different degrees of freedom are given. The relevant part for the reactant HCN and the transition state are shown below:
HCN
Sum of electronic and thermal Enthalpies= -93.240459
Sum of electronic and thermal Free Energies= -93.263374
E (Thermal) CV S
KCal/Mol Cal/Mol-Kelvin Cal/Mol-Kelvin
Total 11.513 6.570 48.229
Electronic 0.000 0.000 0.000
Translational 0.889 2.981 35.816
Rotational 0.592 1.987 11.841
Vibrational 10.032 1.602 0.572
TS
Sum of electronic and thermal Enthalpies= -93.161803
Sum of electronic and thermal Free Energies= -93.186580
E (Thermal) CV S
KCal/Mol Cal/Mol-Kelvin Cal/Mol-Kelvin
Total 8.619 5.971 52.149
Electronic 0.000 0.000 0.000
Translational 0.889 2.981 35.816
Rotational 0.889 2.981 16.331
Vibrational 6.842 0.010 0.001
Notice that translational contributions are identical in the reactant and the transition state because the mass is the same in these two systems. However, rotational and vibrational contributions differ. From the Gibbs free energy values, one can calculate the reaction free energy and the activation free energy:
The free energies of the reaction can be used to calculate the equilibrtium constant. Similarly, the activation free energy can be used with the Eyring-Polanyi equation to calculate the rate constant. The equilibrium and rate constants are temperature dependent, below is a sample calculation at 25 °C:
Clearly, the isomerization of HCN to HNC is too slow, and thermodynamiocally too unfavorable to take place at room temperature.
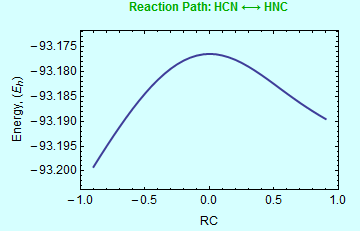
Once the transition state is known, quantum chemistry programs such as Gaussian can follow the reaction path. This "Intrinsic Reaction Path" (IRC) calculation starts from the transition state, and armed with the information about the curvature of the potential energy surface, it will step toward reactants, and then toward products. The calculation is valuable for verifying that the transition state that was found is the one that connects the correct reactants to correct products. The input and output for such IRC calculation, along with a graphical summary of results, is shown below: