

Many bacteria are able to sense the nutrient gradients in the environment and swim toward the highest concentration of the nutrient. Similarly, bacteria can sense poisonous chemicals in their environment and swim away toward the lower concentration of the poison. This phenomenon is called bacterial chemotaxis and involves a cascade of protein-protein interactions that ultimately control the rotational direction of bacterial flagella. In E. coli, the counter-clockwise rotation of flagella causes bacteria to swim straight while the clockwise rotation leads to random tumbling. The chemotaxis occurs because bacteria will swim straight when they are approaching the nutrient source and tumble when they happen to take a direction that takes them away from the food source. The aspartate chemotaxis in E. coli is best understood and structures of several proteins in this signal transduction pathway have been elucidated. However, the molecular details of interactions between the various components of the pathway are unknown.
One key player in the E. coli chemotaxis is the CheY protein. CheY is a 129-residue monomeric soluble protein that regulates the direction of flagellar rotation. The binding of nutrients such as aspartic acid to the aspartate receptor leads to the phosphorylation of a specific aspartate residue in CheY by a kinase, CheA. Phosphorylated CheY binds to the FliM protein in the bacterial flagellar motor, causing the motor to shift to a clockwise rotation and thus inducing tumbling. At the first sight, the interaction of CheY with FliM and CheY appears unlikely because these three proteins are all negatively charged. Specifically, the free CheY has a net charge of about -4 at neutral pH, the CheY-binding domain of CheY (the P2 domain) is predicted to carry a net charge of about -7 and the FliM protein has a charge of -8 at the neutral pH. Despite their similar overall charge, these CheY binds to both CheA and FliM with micromolar affinity.
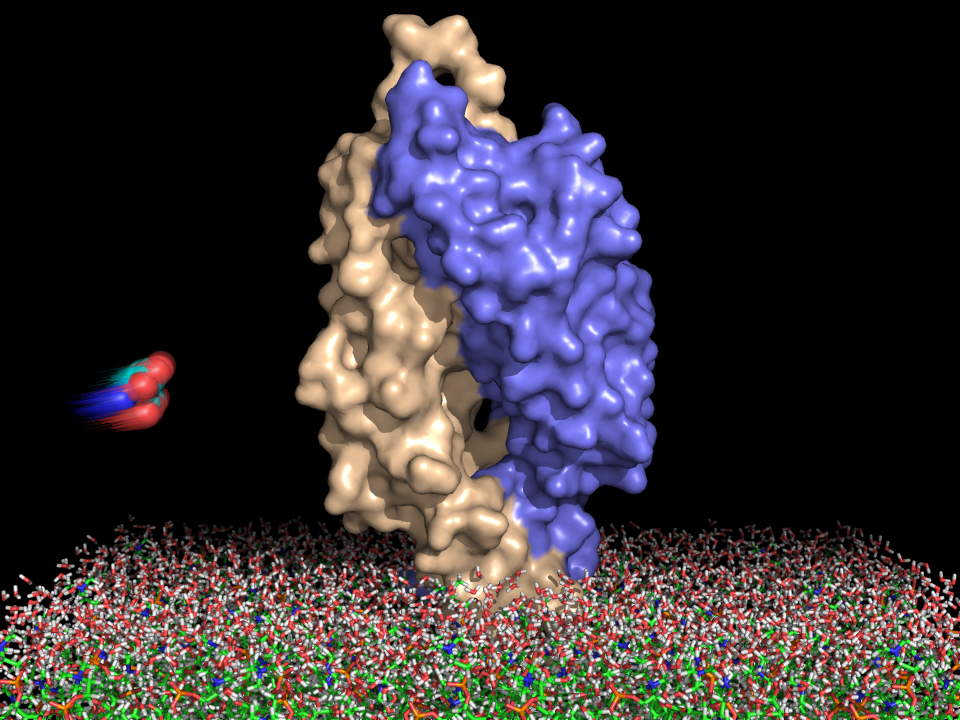
We will now use computer visualization to gain insight into how CheY binds to CheA and and FliM. Such analysis is possible because the crystal structures of the CheY-P2 complex and of the CheY-FliM peptide structures are available. The structure of CheA (the P2 domain) is known from the crystal structure of the complex (PDB code 1a0o). The interaction between CheY and the P2 domain of CheA appears largely electrostatic in nature, which is surprising considering that both CheY and the P2 domain are negatively charged at physiological pH.