

The main findings from these calculations are:
What did we learn about the mechanism of the Zaitsev elimination from these calculations? Can we predict which conformer of the reactant is more reactive? Can we tell if the main product, 1-methylcyclohexene, forms directly from the secondary carbocation or does the secondary carbocation first rearrange to the tertiary carbocation? It turns out that the answer to these questions is not quite obvious from the present set of calculations.
The relative stabilities calculated here are strictly thermodynamic stabilities and cannot be used to answer the question of which conformer is more reactive towards the elimination. In other words, the finding that the conformer with the equatorial methyl group is thermodynamically more stable does not necessarily mean that it must be also kinetically more stable toward the loss of water. The kinetic stability depends on three factors: the ease of protonating the hydroxyl group by the catalytic acid, the thermodynamic stability of the resulting oxonium ion, and the ease of heterolytic breaking of the O-C bond in the oxonium ion.
Strictly speaking, the relative reactivity of the two reactant conformers requires several additional sets of calculations. First, the rate of protonation must be determined. Second, the relative thermodynamic stabilities of the conformers of protonated alcohols must be determined. Third, the reaction barriers for breaking the O-C bond in the two conformers of protonated alcohols must be calculated. However, one can make reasonable guesses about the reactivity without performing any further calculations. First, the rate of proton transfer to the hydroxyl group is expected to be fast and show little dependence of the reactant conformation, unless the base is sterically hindered. Thus, we may well assume that the protonation of alcohol is a fast reversible process, and now the formation of the oxonium ion depends on the basicities of axial and equatorial hydroxyl. There is little reason to think that these basicities differ significantly. Now, the barrier to break the O-C bond becomes a sole determining factor in determining reactivity differences. The principle of the least motion suggests that the elimination of water from the axial position is easier because it involves smaller structural changes than the elimination of water from the equatorial position.
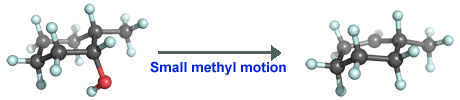
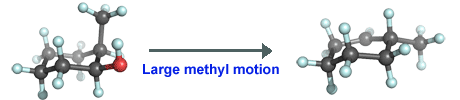
Notice that the image above appears strange because the hydroxyl group in the reactant is in the front but the carbocation center in the product is in the back. One cannot simply rotate this asymmetric carbocation and have the desired structure. However, reflection of this structure gives the desired structure. It sometimes happens that one accidentally performs a calculation on the mirror image of the desired molecule. If this is a problem when making illustrations, one can switch signs of all the coordinates to obtain the mirror image. The two obviously have the same energy (if the tiny effect due to a weak interaction is ignored) and thus there is no need to repeat the expensive calculations on the mirror image. Here, better now:
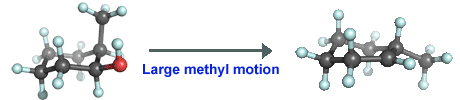
In summary, it seems likely that the most abundant conformer in which the methyl group is equatorial is more reactive as the loss of water from the axial position requires little structural rearrangement of the methyl position.
Strictly speaking, the question whether the secondary carbocation directly deprotonates into 1-methylcyclohexene or rearranges first into the tertiary carbocation requires calculations of the barriers for the proton loss and the 1,2-hydride shift. The former process is difficult to model because in the solution the proton is picked up by a suitable base, such as a cluster of water molecules. The second process is also difficult to model, partially because the accurate description of the hydride transfer requires a basis better than 6-31+G(d,p). Furthermore, there is no guarantee that the MP4(SDQ) method is suitable for description of these two bond breaking and forming processes with the required accuracy. However, one can again make reasonable deductions without carrying out more calculations. Recall that our calculations showed that methylenecyclohexane and 3-methylcyclohexene had nearly identical energies. This means that the ratio of 1-methylcyclohexene to methylenecyclohexane from deprotonation of tertiary carbocation is about the same as the ratio of 1-methylcyclohexene to 3-methylcyclohexene. In other words, if a 50:50 mixture of secondary carbocation and tertiary carbocation deprotonates rapidly, similar amounts of methylenecyclohexane and 3-methylcyclohexene are expected. In the experiment, 3-methylcyclohexene is the main side-product and little methylenecyclohexene is observed, indicating that the secondary carbocation is more likely to deprotonate than undergo the hydride shift. The likely reason for this is a high barrier for the 1,2-hydride shift.
Is summary, it appears that the most likely mechanism for the dehydration of cis 2-methylcuclohexanol involves loss of water from conformer where the methyl group is in equatorial position followed by direct deprotonation of the secondary carbocation into the thermodynamically most stable alkene.
Few things should be kept in mind while attaching chemical significance to these results. First, the calculation was carried out with a fairly small basis. At this level, the energy gap between conformers of the reactant, about 0.9 kcal/mol, may be significantly in error. One common way to estimate the magnitude of this error is to perform a calculation with a somewhat larger basis set, say 6-311+G(df,p) and see how much do the results change. A better way to eliminate the errors due to small basis is to perform calculations with correlation-consistent basis sets, such as cc-pVDZ, cc-pVTZ, and cc-pVQZ and extrapolate the infinite-basis energy using the exponential convergence law. Accurate second order energies can be extrapolated from cc-pVTZ and cc-pVQZ singlet and triplet pair energies.
Second, all the energies that we discussed correspond to the bottom of the energy well but the actual energy of real polyatomic molecules is higher due to bond vibrational levels. Thus, one could make a correction for zero-point vibrational energies based on the result of frequency calculation.
Third, all the structures and energies were obtained in the gas phase but the Zaitsev elimination typically occurs in condensed medium. Polar solvents usually stabilize the structure with a higher dipole moment. Inspection of the optimization output files reveals that the dipole moment of the two conformers of the reactant are very similar (2.01 D for equatorial methyl, 1.95 D for axial methyl conformer). These differences are not too significant; in practice one could carry out a calculation in the presence of a solvent model to estimate the difference in solvation of the two conformers. Similarly, the relative stabilities of alkenes are slightly solvent dependent. In this case, methylencecyclohexene has the largest dipole moment (0.70 D) is expected to be most stabilized by the solvent. The estimation of solvent effects on relative stability of the two carbocations is more challenging because these molecules carry a net charge.